

Si profila oggi la possibilità di correggere direttamente il DNA genico tagliandone il frammento mutato e sostituendolo con la sequenza normale. È il sistema CRISPR/Cas9. Una prospettiva al momento ancora ai primi passi, ma potenzialmente in grado di curare le malattie genetiche, compresa la fibrosi cistica.
Nell’era della medicina personalizzata per la fibrosi cistica (FC), con l’uso in clinica dei cosiddetti correttori e potenziatori, la terapia genica sta avendo una rinascita. Non tanto a livello clinico, in cui l’unico gruppo attivo, lo UK Cystic Fibrosis Gene Therapy Consortium, ha prodotto dei dati di bassa efficacia sull’aerosolizzazione di vettori non virali veicolanti il gene wild-type CFTR [1], ma a livello di ricerca preclinica.
Com’è noto, la terapia genica può avvalersi di due possibili strategie per correggere il gene CFTR mutato: una di gene adding, ovvero di aggiunta del gene wild-type (cioé con la sequenza normale), mediante vettori virali e non virali, nelle cellule target dell’epitelio respiratorio bronchiale/bronchiolare; l’altra di gene editing, cioè di correzione in situ (a livello del cromosoma 7, dove si trova il gene CFTR), solitamente mediante vettori non virali. Sulla prima sono stati condotti studi estensivi fin dalla clonazione del gene (1989), ma con i risultati poco esaltanti accennati prima. D’altra parte, la seconda ha utilizzato sistemi di correzione poco efficienti, fino alla scoperta di un sistema composto da acidi nucleici (CRISPR) ed enzimi del tipo nucleasi, detto CRISPR/Cas9. Esso è stato primariamente identificato come un sistema difensivo dei procarioti (batteri) contro organismi patogeni invasori (come i virus), in quanto permette di riconoscere e degradare gli acidi nucleici di questi ultimi (quindi fa parte del sistema immune adattativo ancestrale).
In breve, la nucleasi Cas9 è un enzima capace di tagliare il DNA del genoma in una maniera sequenza-specifica definita da un RNA guida, che è complementare (lo riconosce e vi si appaia) al segmento di DNA da tagliare. L’enzima Cas9 e l’RNA guida sono il sistema che i batteri usano per eliminare i virus che li attaccano (speciali virus detti batteriofagi); viene utilizzato in modo da tagliare, incorporare e depositare nel genoma batterico il DNA virale (che va a costituire le sequenze CRISPR) e dotare così il batterio di una memoria dei geni dei virus eliminati. In seguito a ulteriori incontri, il virus scatena la memoria del batterio che così produce la nucleasi Cas9 e l’RNA guida, che degradano il DNA del virus e lo neutralizzano.
In chiave figurata possiamo immaginare la nucleasi Cas9 come una forbice e l’RNA guida come l’elemento che dirige la forbice sul tratto di DNA da tagliare. Una volta conosciuto il sistema CRISPR/Cas9 e il suo modo di funzionare, si è visto che gli RNA guida possono essere costruiti anche in laboratorio, conoscendo la sequenza di DNA che devono catturare. Sfruttando la capacità del sistema CRISPR/Cas9 di effettuare precise rotture nella doppia elica del DNA, si è aperta la strada alla strategia di gene editing (modifica dei geni). Difatti, CRISPR/Cas9 può essere usato dalla comunità medica al fine di correggere le mutazioni causanti malattia, grazie alla scoperta che CRISPR/Cas9 può lavorare in tandem con sistemi di riparazione cellulari (cellule staminali) in modo da inserire delle delezioni o inserzioni che possono spegnere un gene (cosiddetto knock-out) o riparare un gene in situ (cosidetto knock-in). In quest’ultimo caso, insieme a Cas9 e all’RNA guida, si aggiunge alla cellula da modificare un frammento di DNA che viene integrato al posto della parte che è stata prima rimossa, sfruttando un meccanismo cellulare che si chiama ricombinazione omologa (HR) [2].
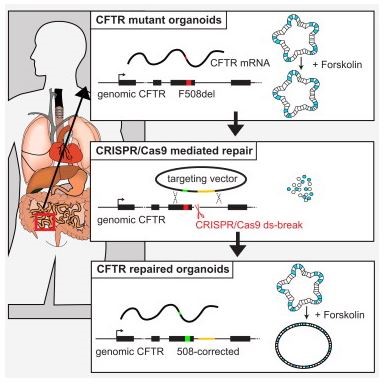
Figura dalla referenza [3],
www.cell.com/cell-stem-cell/abstract/S1934-5909(13)00493-1?_returnURL=http%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS1934590913004931%3Fshowall%3Dtrue.
Negli organoidi ottenuti da cellule staminali intestinali di individui omozigoti per la mutazione F508del, lo stimolo della CFTR (ottenuto attraverso una sostanza chiamata forskolin) non può attivare la proteina mutata ed essi non si rigonfiano (pannello superiore). In seguito all’inserimento all’interno delle cellule staminali del sistema CRISPR/Cas9 (che induce la rottura del doppio filamento di DNA ed è chiamato ds-breaks) e del vettore di DNA che possiede la sequenza wild-type (targeting vector, pannello intermedio), si ha la correzione della mutazione e gli organoidi così generati possono rispondere a forskolin aumentando di volume (pannello inferiore), dimostrando che essi esprimono una proteina CFTR funzionante.
La ricombinazione omologa è un meccanismo di riparazione del DNA basato sulla possibilità di scambiare materiale genetico tra frammenti di DNA che possiedono ampie regioni uguali o molto simili. Viene usata dalla cellula per riparare il DNA che presenti rotture di entrambi i suoi due filamenti, a differenza di altri meccanismi di riparazione ha il vantaggio di non introdurre nuove mutazioni, ed è risultata fondamentale per le applicazioni mediche ai fini terapeutici di malattie geneticamente determinate, inclusa la fibrosi cistica. Come illustrato nella figura, dopo che il filamento di DNA contenente la mutazione CFTR in questione (in questo caso la mutazione F508del) è stato artificialmente rotto e tagliato per effetto di CRISPR/cas9, la rottura viene riparata con un frammento simile ma contenente la corretta sequenza di DNA.
La prima dimostrazione delle potenzialità di CRISPR/Cas9 in fibrosi cistica è arrivata nel 2013, quando il gruppo di Utrecht in Olanda ha usato tale sistema per correggere il gene CFTR con la mutazione F508del in un modello di organoidi intestinali ottenuti da cellule staminali adulte, isolate da due pazienti pediatrici omozigoti per la mutazione [3].
Gli organoidi rappresentano un modello tridimensionale di epitelio del piccolo intestino, quindi più vicino a una situazione in vivo dal punto di vista strutturale, ma non solo: posseggono anche delle proprietà consequenziali alla presenza di una proteina CFTR funzionante, ovvero si rigonfiano, in seguito alla secrezione di cloro e di acqua, mediante opportune stimolazioni. Tali organoidi sono stati transfettati con (ovvero si fa entrare al loro interno) un frammento di DNA wild-type per CFTR insieme a CRISPR/Cas9, ottenendo un comportamento degli organoidi simile a quello di organoidi ottenuti da soggetti non-FC (vedi figura). Inoltre, l’analisi genomica identificava proprio nella correzione sito-specifica la causa di questo effetto terapeutico.
Come si può facilmente intuire, questi interventi di terapia genica mediante CRISPR/Cas9 sono facilmente adattabili a ogni mutazione, perché basta cambiare le sequenze di RNA guida che devono appaiarsi col DNA genomico dove risiede la mutazione, ponendo veramente le basi per quella che può essere chiamata medicina di precisione o medicina personalizzata. Resta da studiare se tale tecnica molto potente possa essere applicata alla correzione della malattia respiratoria. Una linea di ricerca promettente sembra essere quella di coniugare CRISPR/Cas9 con le cellule staminali pluripotenti indotte (iPSC). Queste ultime sono una nuova frontiera nell’ambito dell’utilizzo terapeutico di cellule staminali, in quanto vengono prodotte da cellule differenziate adulte dello stesso paziente (ad es. facilmente ottenibili, come i fibroblasti della pelle) mediante una loro riprogrammazione in vitro e, in seguito alla correzione della mutazione con interventi di terapia genica, eventualmente reintrodotte nello stesso paziente, senza quindi alcuna possibilità di un rigetto.
I ricercatori hanno quindi isolato dei fibroblasti cutanei da un paziente FC F508del, che sono stati riprogrammati a iPSC e quindi trattati con CRISPR/Cas9 specifici per il sito genomico del CFTR e un vettore con una sequenza wild-type per CFTR (in effetti tutti questi elementi vengono transfettati nel nucleo delle iPSC) [4]. Quello che è interessante è che le iPSC così corrette sono state indotte a differenziarsi in cellule epiteliali respiratorie, le quali hanno mostrato una proteina CFTR correttamente trasportata sulla membrana e funzionante. Nessuna mutazione aggiuntiva è stata riscontrata, mostrando la sicurezza di questa strategia. Quindi, in linea di principio, potrebbe essere possibile ottenere delle iPSC da cellule del polmone di un paziente FC, trattarle con CRISPR/Cas9 per correggere la mutazione, e reinserirle nel polmone in una di quelle nicchie dove le cellule staminali trovano il microambiente adatto alla loro sopravvivenza e crescita. Sono state identificate molte nicchie staminali nel polmone, la più promettente delle quali sembra essere quella che dà ospitalità alle cosiddette cellule staminali broncoalveolari.
In alternativa, CRISPR/Cas9 dovrebbe essere veicolato all’epitelio respiratorio mutato mediante vettori tradizionali di terapia genica. I vettori virali sembrano inappropriati a questo fine, a causa delle discrete dimensioni del sistema CRISPR/Cas9 che deve essere integrato nel genoma virale per essere trasportato all’interno delle cellule; i vettori non virali (lipidici o polimerici) sembrano più adatti, anche perché essi non si integrano nel genoma dell’ospite e quindi sono esenti dal rischio di possibile induzione di tumori secondari [5]. Qualora somministrati per aerosol, non sappiamo però se sono capaci di superare la formidabile barriera rappresentata dal muco patologico denso e viscoso che sovrasta l’epitelio target. In ogni caso la via di somministrazione elettiva dovrà essere quella locale (tramite aerosol), poiché è stato dimostrato da vari gruppi di ricerca che la via sistemica (somministrazione endovenosa), per ragioni anatomiche, raggiunge la zona alveolare del polmone, sede degli scambi gassosi, e non quella della sede di malattia FC, ovvero l’albero bronchiale/bronchiolare.
1. Alton EW, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. Repeated nebulisation ofnon-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double
blind, placebocontrolled,phase 2b trial. Lancet RespirMed. 2015 Sep;3(9):684-91.
2. Charpentier E., Kaldy P. L’enzima che rivoluziona la genetica. Le Scienze 2016; 572: 29-
35. (articolo divulgativo che illustra la storia della scoperta di CRISPR/Cas9 e le sue possibili
applicazioni)
3. Schwank G, Koo B-K, Sasselli V, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13(6):653–658.
4. Firth AL, Menon T, Parker GS, et al. Functional gene correction forcystic fibrosis in lung epithelial cells generated from patient iPSCs.Cell Rep. 2015;12(9):1385–1390.
5. Li L, He Z-Y, Wei X-W, et al. Challenges in CRISPR/CAS9 delivery: potential roles of nonviral vectors. Hum Gene Ther. 2015;26(7):452–462.