Chi scrive probabilmente vuole sapere quali sono le possibilità che la procedura dello screening neonatale, basata su un insieme di accertamenti (di cui il principale è il valore dell’IRT=tripsinogeno immunoreattivo), dia origine a sospetti infondati di fibrosi cistica, cioè casi chiamati “falsi positivi”. La letteratura scientifica mostra che il numero di falsi positivi allo screening neonatale per FC è variabile, ma comunque basso fino a bassissimo se all’IRT viene associata l’indagine genetica (ne abbiamo parlato qui e qui). Infatti è estremamente raro che il test diagnostichi nel neonato la presenza di mutazioni del gene CFTR che in realtà non ci sono; semmai è più frequente che sfuggano mutazioni che invece ci sono. Il test del sudore rappresenta l’accertamento diagnostico definitivo al termine della procedura di screening (qui un approfondimento).
Nel caso di questo bambina il risultato del test del sudore si può interpretare come in accordo con la presenza delle due mutazioni G1244E e D1152H, che sono mutazioni particolari. Per informazioni dettagliate si possono leggere due precedenti risposte: questa (su G1244E) e questa (su D1152H).
G1244E è mutazione cha appartiene al gruppo delle mutazioni di gating, D1152H al gruppo delle mutazioni con funzione residua. Molto sinteticamente si può dire che entrambe non danneggiano gravemente la sintesi della proteina CFTR, che agisce come canale del cloro. Per questo si associano per lo più a livelli di cloro nel sudore non particolarmente elevati oppure nella fascia incerta (“borderline”). Si sa anche che c’è in genere correlazione fra la presenza di mutazioni che non provocano danno grave sulla proteina CFTR e sintomi più modesti e andamento più benigno della malattia FC. Questo ovviamente in linea generale, le previsioni a livello individuale sono molto difficili.
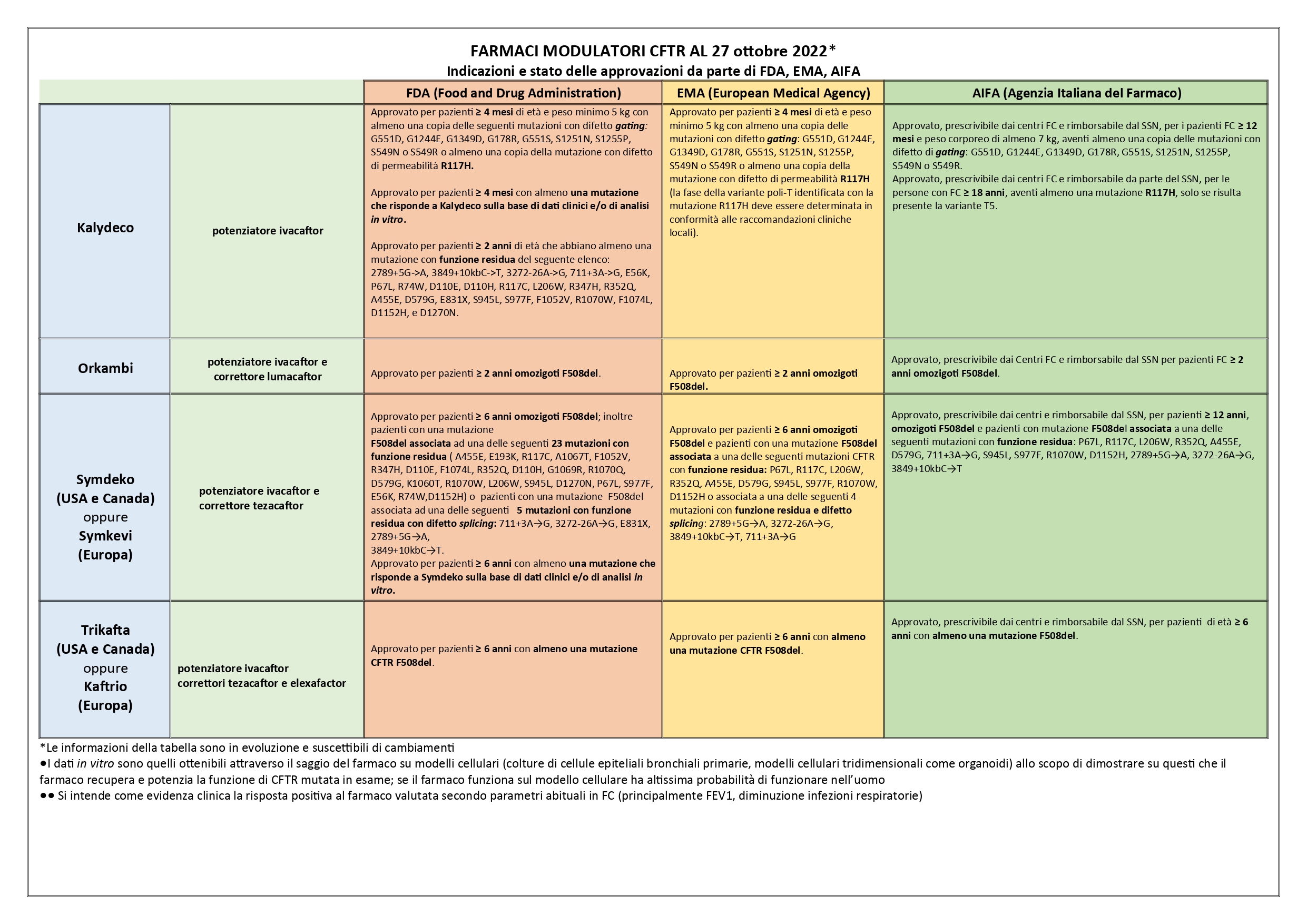
L’informazione importante è che la mutazione G1244E è trattabile con il farmaco Kalydeco (potenziatore della proteina CFTR), fornito a carico dal SSN e prescrivibile su valutazione del centro di cura a partire dall’infanzia; la mutazione D1152H è anch’essa trattabile con Kalydeco, però il farmaco, approvato da FDA negli USA, non è stato ancora approvato in Europa dall’ente regolatore EMA (qui un riassunto sulle indicazioni e lo stato delle approvazioni di FDA, EMA e AIFA per i farmaci modulatori di CFTR).
La possibilità di un trattamento farmacologico anche per una sola delle due mutazioni CFTR presenti nel genotipo determina un radicale miglioramento della qualità e della durata della vita dei bambini che nascono oggi con fibrosi cistica.

{kind=link}