Sappiamo da studi in vitro che combinando un correttore (Lumacaftor) con un potenziatore (Ivacaftor) si ottiene un recupero di funzione del canale del cloro CFTR difettoso a causa della più diffusa mutazione, la F508del. E’ stato condotto anche uno studio clinico di fase 2 con la stessa combinazione, di cui sono stati forniti succinti commenti su questo sito in Progressi di ricerca del 23.12.13 (Nuovo commento agli studi clinici di fase 2 con Lumacaftor (VX-809) combinato a Ivacaftor (VX-770, kalydeco): su quello studio è uscita recentemente una pubblicazione (1). Alla base di questo approccio terapeutico sta la nozione che la proteina CFTR mutata per DF508 viene intrappolata nel citoplasma della cellula e viene quindi rimossa prima di subire adeguata maturazione e quindi trasporto alla membrana cellulare sua sede di azione. Il correttore Lumacaftor sarebbe in grado di recuperare almeno in parte la proteina e di favorirne la maturazione e il trasporto in membrana: l’intervento abbinato del potenziatore Ivacaftor otterrebbe dalla proteina, pur difettosa ma giunta in membrana, uno stimolo importante al recupero anche della sua funzione di trasportare cloro.
Sono stati resi noti in questi giorni, attraverso un comunicato stampa della Vertex, i risultati di due studi (TRAFFIC E TRANSPORT) randomizzati di fase 3, in doppio cieco, controllati con placebo, disegnati per valutare l’efficacia e la sicurezza di Lumacaftor (correttore) in combinazione con Ivacaftor (potenziatore) nei pazienti CF con età di 12 anni e oltre, omozigoti per la mutazione F508del. I due studi, organizzati e supportati dalla compagnia Vertex Pharmaceuticals, hanno un protocollo identico, in tutti i particolari. Cambia solo il numero di registrazione, un outcome in più in uno dei due studi (ECG in ambito ambulatoriale), e i paesi in cui vengono condotti (TRAFFIC: United States, Australia, Canada, Czech Republic, France, Germany, Ireland, Italy, Netherlands, Sweden, United Kingdom; TRANSPORT: United States, Australia, Austria, Belgium, Canada, Denmark, France, Germany, Spain, United Kingdom).
Ecco i link ai protocolli
clinicaltrials.gov/ct2/show/record/NCT01807923
clinicaltrials.gov/ct2/show/record/NCT01807949
Si riporta qui la traduzione italiana del comunicato, con leggere modifiche, e di seguito un nostro commento.
Ognuno dei 2 studi ha reclutato due gruppi di trattamento attivo e un gruppo placebo.
I gruppi di trattamento attivo:
· lumacaftor 600 mg una volta al giorno + ivacaftor 250 mg ogni 12 ore;
· lumacaftor 400 mg ogni 12 ore + ivacaftor 250 mg ogni 12 ore. I due dosaggi di Lumacaftor sono stati scelti sulla base dei risultati del già citato studio di fase 2 (1).
I pazienti nello studio hanno continuato i loro trattamenti in corso con le terapie standard.
1.108 soggetti in tutto hanno ricevuto almeno una somministrazione del farmaco, suddivisi nei due studi (549 TRAFFIC, 559 TRANSPORT) in circa 200 centri di sperimentazione clinica in tutto il Nord America, Europa e Australia.
L’esito (outcome) primario degli studi è stata la variazione assoluta, rispetto al basale, del FEV1 (valore % del predetto teorico) al termine del periodo di trattamento di 24 settimane; l’analisi statistica è stata eseguita con un modello misto per Misure ripetute (MMRM).
Tra gli obiettivi secondari, il numero di infezioni respiratorie, l’indice di massa corporea per la valutazione dello stato nutrizionale, la valutazione della qualità di vita relativamente alla situazione respiratoria.
Sulla base del disegno dello studio, che comprendeva diversi bracci di trattamento all’interno di ogni studio, la significatività statistica per ciascun braccio contro placebo era basata su un valore di p < o = a 0.025. Oltre alle analisi per ciascuno dei due studio, viene presentata una analisi congiunta (pre-pianificata) dei 2 studi.
Efficacia – Risultati – Funzione polmonare (ppFEV1):
Il valore medio di partenza del FEV1 era pari circa al 61 per cento del predetto (per tutti i pazienti). Tutti e quattro i gruppi di trattamento attivo, nell’ambito dei due studi, hanno mostrato un effetto significativo del trattamento sull’esito primario. Inoltre sono stati osservati miglioramenti assoluti e relativi medi della funzione polmonare statisticamente significativi per tutti e quattro i gruppi di trattamento, sia entro gruppo che contro placebo, in tutte le rilevazioni all’interno dello studio (settimane 2, 4, 8, 16 e 24).
I risultati dettagliati di ciascun braccio dello studio sono forniti qui di seguito:
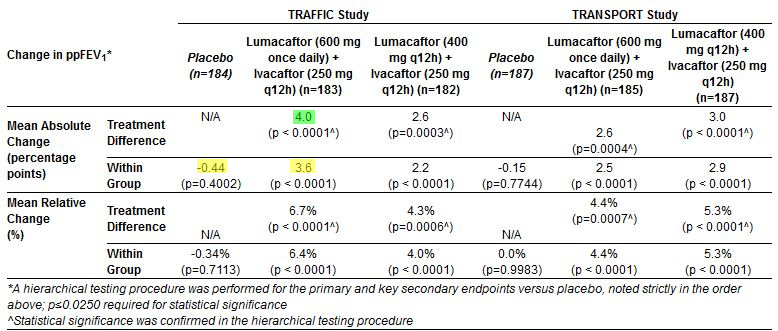
Guida alla lettura delle tabelle, per chi non fosse abituato: in ogni tabella la seconda riga presenta le variazioni entro gruppo: nella tabella sopra, il gruppo placebo peggiora di 0,4 mentre il gruppo trattato migliora di 3,6, perciò l’effetto TRA gruppi è la differenza 3,6 – (-0,4) = 4,0
Per una migliore comprensione dei termini Variazione Assoluta (absolute change) e Variazione Relativa (relative change): la variazione assoluta è la differenza tra due valori; quella relativa è la stessa differenza, ma rapportata al valore iniziale. Nel nostro caso purtroppo la cosa è ancor più complicata dal fatto che il valore di FEV1 è già un percento rispetto al previsto in base al sesso e all’età. In ogni caso, trascurando questo dettaglio, supponiamo che un paziente passi da 60 a 63 di punti percentuali di FEV1: la variazione assoluta in questo caso vale 3 (63-60) mentre quella relativa è 5%: (63-60)/60.
Efficacia. Risultati – endpoint secondari:
All’interno dello studio TRAFFIC, le persone che hanno ricevuto i due regimi di combinazione hanno avuto, rispetto al placebo, una riduzione del 28 (RR=0,72) e del 34 (RR=0.66) per cento delle esacerbazioni polmonari (peggioramento sintomatologico della malattia che richiede un trattamento con antibiotici) durante il periodo di trattamento di 24 settimane. Analogamente, nel TRANSPORT, riduzione del 31 (RR=0,69) e 43 (RR=0,57) per cento.
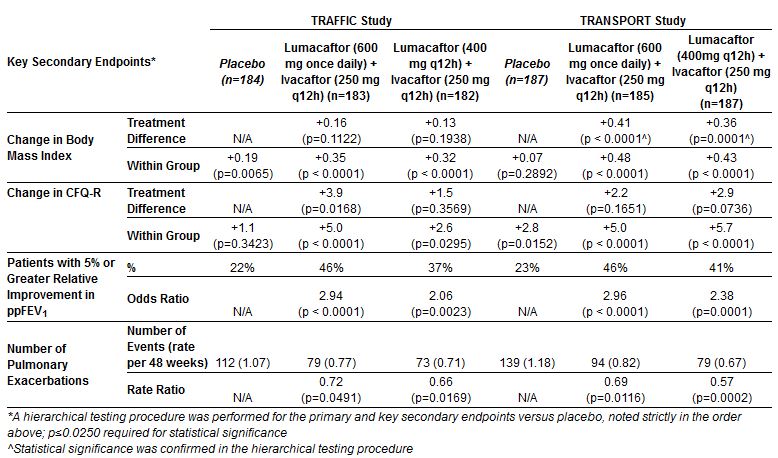
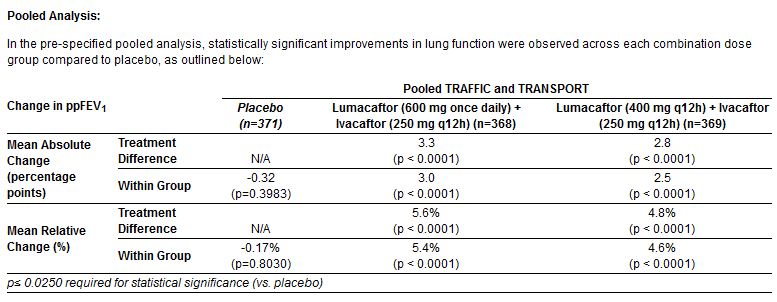
Analisi congiunta dei due studi:
Esito primario
Esiti secondari
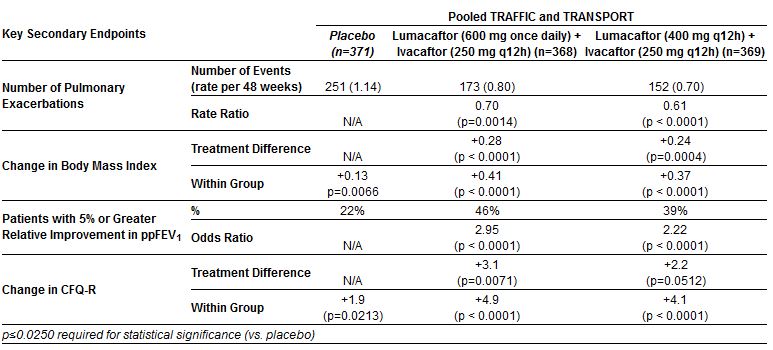
Come si vede dalla lettura delle tabelle, l’analisi congiunta dei due studi (tutti i casi messi insieme) conferma quanto già mostrato dalle tabelle precedenti, con un aumento assoluto del 3,3% e 2,8% del FEV1 medio nei due gruppi di trattamento rispetto al placebo. E così pure con la riduzione dei tassi di esacerbazione polmonare, rispetto a quelli che hanno ricevuto placebo, del 30 (RR=0,70) e 39 (RR=0,61) per cento. Miglioramenti statisticamente significativi, seppur modesti, anche dell’indice di massa corporea e della qualità della vita (per l’aspetto respiratorio) misurata con lo strumento CFQ-R.
Sicurezza:
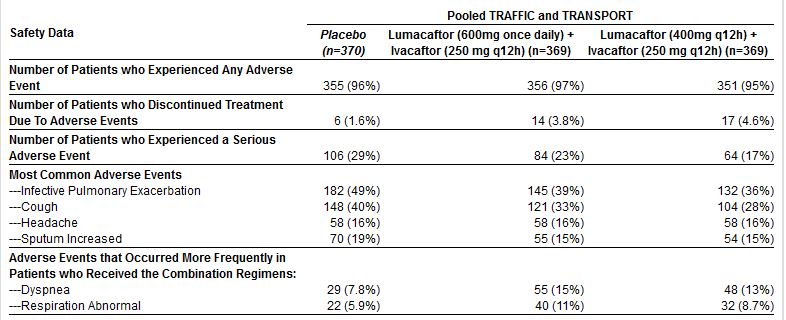
I risultati sulla sicurezza di questi studi sono presentati in forma congiunta per i due studi. I farmaci sono stati generalmente ben tollerati. Gli eventi avversi più comuni, indipendentemente dal gruppo di trattamento, sono stati: riacutizzazione infettiva polmonare, tosse, mal di testa e aumento dell’espettorato; si sono verificati più frequentemente nei pazienti trattati rispetto a quelli che avevano ricevuto il placebo.
Il 4,2 per cento di tutti i pazienti trattati, indipendentemente dal gruppo di dosaggio, ha interrotto il trattamento a causa di eventi avversi, rispetto al 1,6 per cento del gruppo placebo. L’aumento degli enzimi epatici (superiore a tre volte il limite superiore della norma) sono stati osservati nel 5,2 per cento dei trattati rispetto al 5,1 per cento del gruppo placebo.
Sette pazienti trattati hanno avvertito gli eventi avversi importanti legati alle anomalie dei test di funzionalità epatica, rispetto a nessun paziente del gruppo placebo. Dopo la sospensione o interruzione del trattamento di combinazione, i test sono tornati ai valori basali per sei dei sette pazienti, e il settimo è migliorato sostanzialmente.
Tutti i pazienti che hanno completato lo studio di 24 settimane, indipendentemente dal trattamento assegnato, hanno avuto la possibilità di iscriversi in uno studio di rollover. Dopo la fine del periodo di somministrazione di 24 settimane, più di 1.000 pazienti hanno scelto di entrare nello studio di rollover e continuare a ricevere la terapia di combinazione.
Commento a questi dati preliminari, in attesa dell’articolo scientifico completo
I risultati dei due studi sembrano discretamente incoraggianti, ma vanno ovviamente confermati su un numero ancora maggiore di pazienti e su un più lungo periodo.
Dispiace che nel protocollo degli studi non sia stato previsto un indice di miglioramento del difetto di base (es cloro nel sudore), che avrebbe irrobustito i risultati, dando loro anche un razionale più convincente.
E’ abbastanza strano che siano stati lanciati in contemporanea due studi identici, e non un solo grande studio: non sono riuscito a trovare spiegazioni di questa scelta.
I pazienti previsti erano in totale 1122 (in circa 200 centri: circa 5 in media per centro). Il comunicato Vertex presenta i dati di 1108 soggetti, che hanno ricevuto almeno una somministrazione del farmaco. Mancherebbero i dettagli sui 14 mancanti (non arruolati? Non randomizzati? Persi al follow up?).
Un dato confortante a favore dell’efficacia del trattamento è la diminuzione del tasso di esacerbazioni respiratorie, che passa da 1,14 (casi per paziente all’anno, più precisamente, proiettati su 48 settimane) con il placebo a 0,8 e 0,7 con la terapia combinata. E anche lo stato nutrizionale sembra avere un certo miglioramento, anche se lieve.
Per contro va osservato come, nel gruppo placebo, a fronte di un andamento medio verso il peggioramento del FEV1 (variazione relativa -0,17%, analisi congiunta), ben il 22% dei soggetti trattati mostri un miglioramento maggiore o eguale al 5% (rispetto al 46% e al 39% dei due trattamenti). In assenza di dati sulla variabilità (vengono infatti presentati solo i valori medi e non le deviazioni standard), ci sembra di capire che questa sia molto ampia. Sarebbe importante capire bene quanti pazienti alla fine ricevono beneficio, e di quale entità: dal grande beneficio, al beneficio scarso, al peggioramento.
Infine non è possibile capire da questi dati come si possa interpretare il punteggio che misura la qualità del benessere respiratorio (percepito dal paziente), risultato significativo per uno dei due bracci nell’analisi congiunta.
Alcuni dettagli sugli effetti avversi importanti (“serious”) non sono chiari: si dice che sono più frequenti nel gruppo placebo: 29% (!) vs 23% e 17% nei trattati; e tuttavia gli eventi che portano alla sospensione della terapia sono più frequenti nei trattati (3,8% e 4,6% dei pazienti) rispetto al gruppo placebo (1,6%). Sarebbe utile avere maggiori dettagli su quali eventi siano stati considerati.
Molti interrogativi ancora aperti dunque, che autorizzano a pensare che la combinazione ideale di correttore e potenziatore debba essere ancora molto ricercata, come molti gruppi di ricerca stanno facendo. Naturalmente tutte le valutazioni andranno rivedute quando sarà disponibile il report completo e, in futuro, quando le variazioni osservate saranno estese a gruppi di pazienti ancora più ampi e per periodi più lunghi.
1. Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, Huang X, Waltz D, Patel NR, Rodman D; on behalf of the VX09-809-102 study group. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014 Jun 24. pii: S2213-2600(14)70132-8. doi: 10.1016/S2213-2600(14)70132-8. [Epub ahead of print]